一.测试服务器设定
1.使登录后自动进入/home目录下
1 | vim ~/.bashrc |
2.新建RNAseq_tool文件夹,存放各工具
1 | mkdir RNAseq_tool |
二.各生信工具在测试服务器下的安装
1.sratoolkit下载及安装
1 | #下载并解压 |
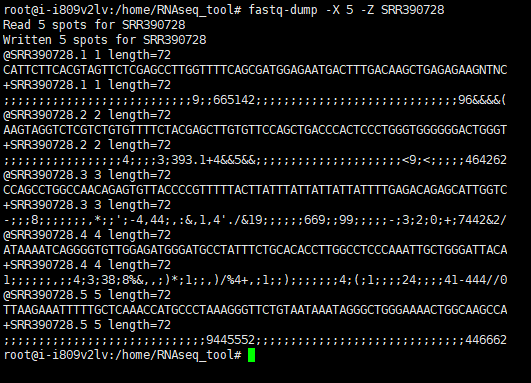
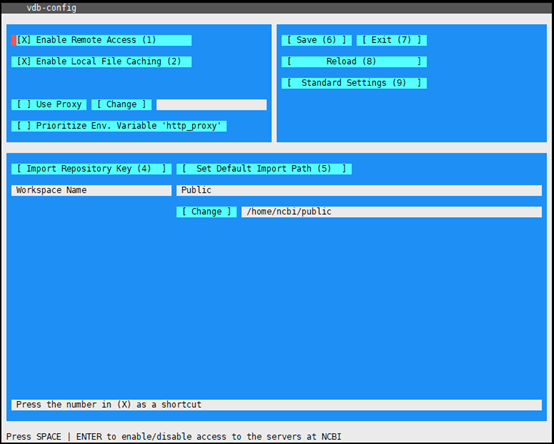
1 | #配置SRAtoolkit的下载路径 |
2.fastqc下载及安装
1 | #fastqc需要java环境,首先下载并配置java,此处下载 jre1.8.0_144 |
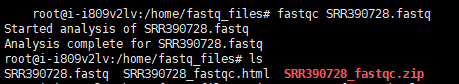
处理好的SRR390728_fastqc.html可以通过WinSCP下载到Windows系统上进行查看。
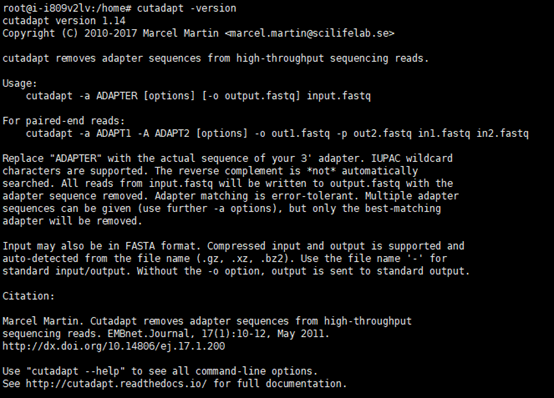
3.cutadapt下载及安装
1 | #安装python-pip,python-dev |
4.fastx-toolkit下载及安装
1 | #下载并安装依赖库libgtextutils 0.7 |
5.安装bowtie2(旧流程)
1 | #安装依赖包libtbb2 |
6.安装Tophat2(旧流程)
1 | #下载并解压 |
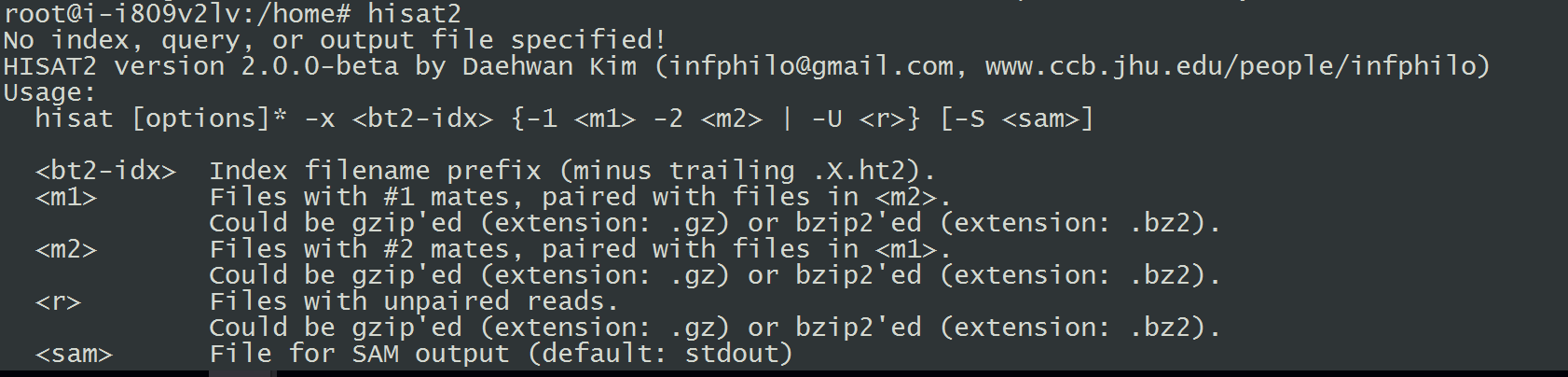
7.安装HISAT2(Tophat替代工具)
1 | #下载源码并解压 |
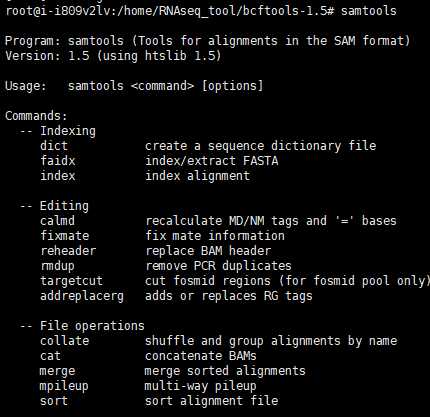
8.安装samtools
1 | #下载依赖库:libncurses5-dev, zlib1g-dev libbz2-dev liblzma-dev |
9.安装RSeQC(用于对bam文件进行质控,项目主页:http://rseqc.sourceforge.net/)
1 | # Python2.7环境下 |
其由许多功能脚本组成,具体可以看官网信息(http://rseqc.sourceforge.net/)
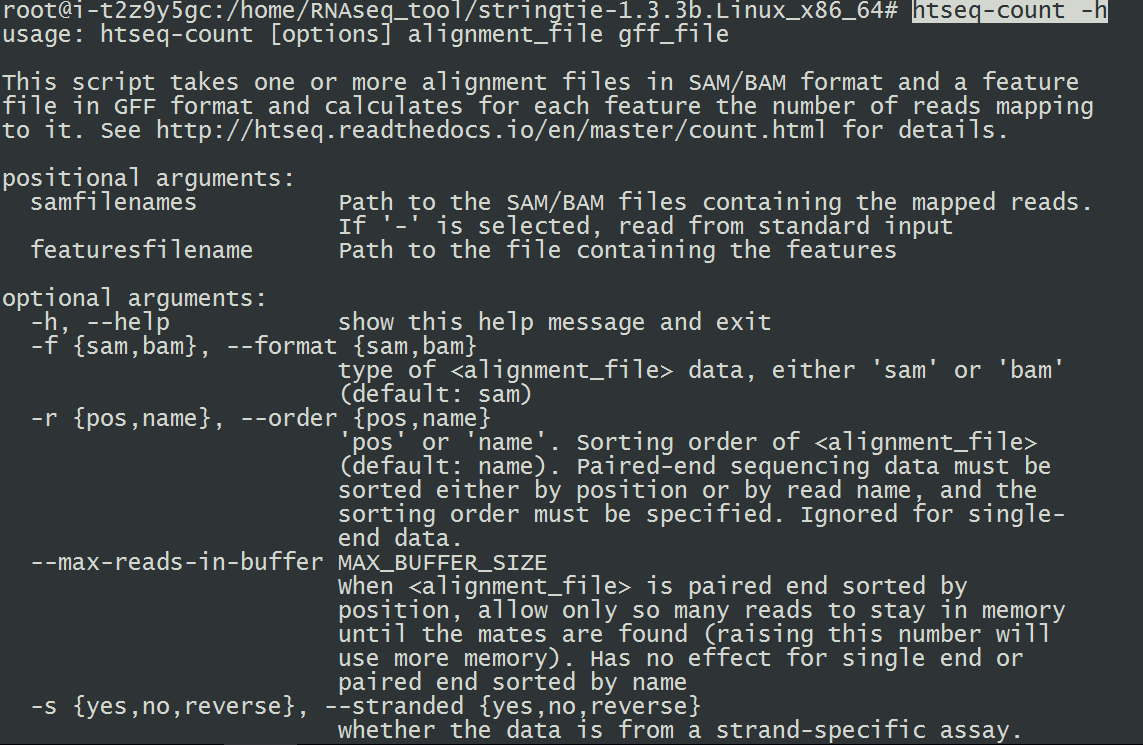
10.安装HTseq(项目主页:http://htseq.readthedocs.io/en/release_0.9.1/)
1 | # 安装依赖 |
11.安装bedtools
1 | #下载并解压 |

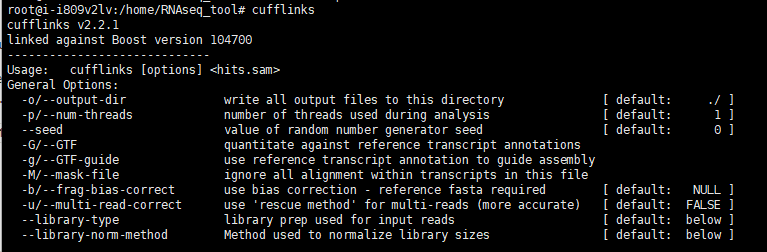
12.安装cufflinks(旧流程)
1 | #下载并解压 |
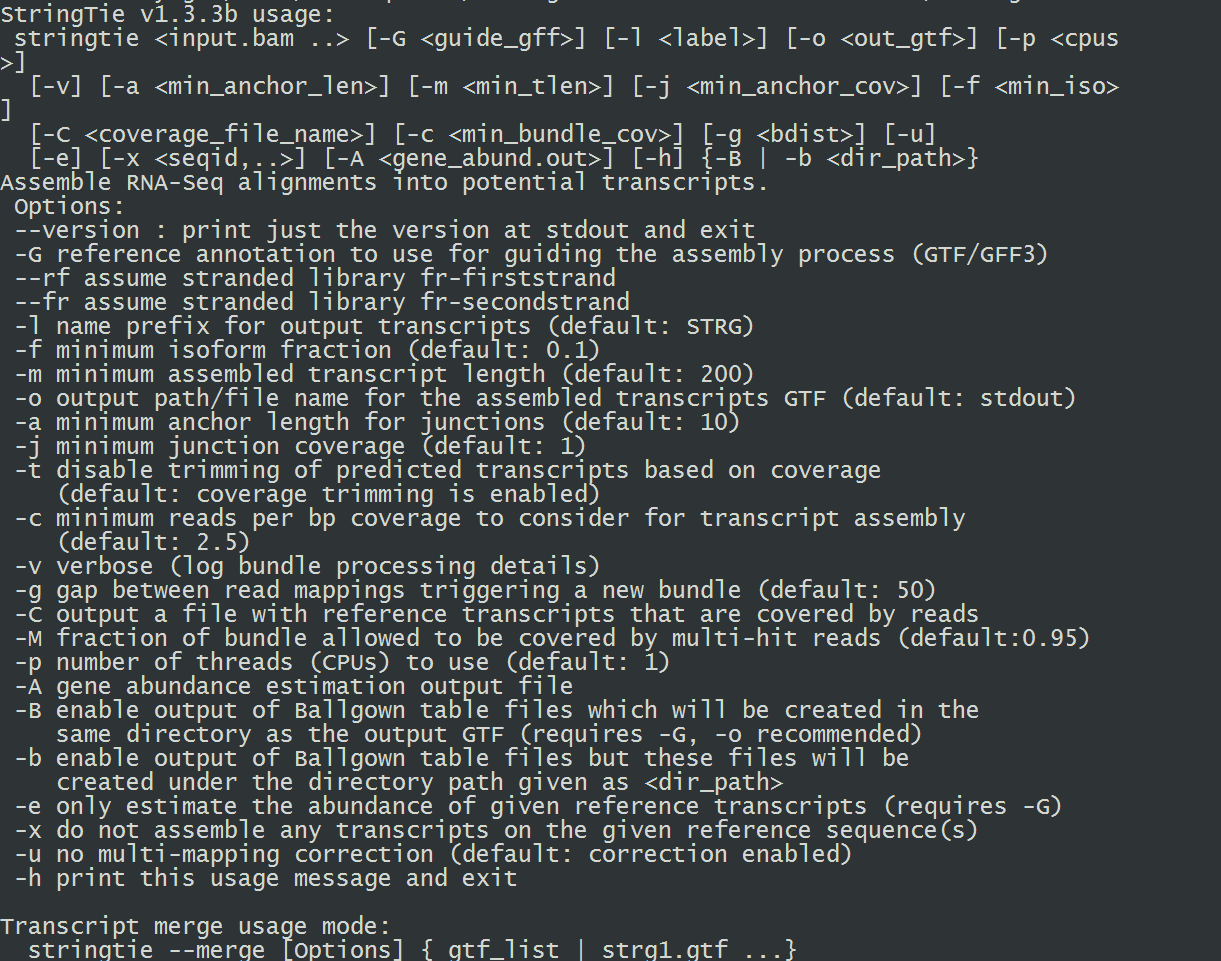
13.安装stringtie(cufflinks替代文件,项目主页在此)
1 | #下载并解压 |